

contenuti
Indice dei contenuti
- Cosa sono i Dispositivi Medici (DM)
- La vigilanza sui DM
- Il sistema di vigilanza e la rete nazionale di vigilanza
- La rete regionale di vigilanza
- I responsabili della vigilanza sui DM
- Cosa, come e quando segnalare
- Protocollo regionale per lo svolgimento delle attività di vigilanza sui dispositivi medici e i dispositivi medici in vitro
Cosa sono i Dispositivi Medici (DM)
I dispositivi medici sono una gamma molto ampia di prodotti e tecnologie dalle caratteristiche
diverse, utilizzati nelle strutture sanitarie per la diagnosi e l’assistenza, o direttamente dal
paziente a domicilio, nell’ambito dei percorsi di cura (per esempio siringhe, cateteri, stomie,
protesi impiantabili, ferri chirurgici, apparecchiature sanitarie).
La rilevanza dei DM in ambito sanitario è correlata al loro ampio utilizzo in tutti i
processi assistenziali e al crescente impegno economico correlato al loro uso.
Trattandosi di prodotti che, una volta certificati secondo quanto previsto dalla normativa,
non necessitano di un’autorizzazione da parte dell’autorità competente per poter essere immessi nel
mercato, sono particolarmente importanti le attività di vigilanza e controllo attuate per
monitorare la loro sicurezza e affidabilità.
La vigilanza sui DM
Negli ultimi anni l’Unione Europea ha riformulato il quadro normativo che regola i dispositivi medici con la finalità di assicurare a livello della comunità europea un quadro regolatorio basato sulla solidità, la trasparenza, la sostenibilità e la garanzia di un elevato livello di sicurezza supportato, al contempo, dall’innovazione tecnologica.
Sono stati così elaborati il Regolamento Europeo 745/2017 relativo ai dispositivi medici ed il Regolamento Europeo 746/2017 relativo ai dispositivi medico-diagnostici in vitro, che sostituiranno completamente le precedenti direttive europee.
I regolamenti annoverano la vigilanza tra gli elementi chiave dell'attuale approccio normativo. Infatti le attività di vigilanza sui DM garantiscono il controllo ed il monitoraggio del prodotto durante tutto il suo ciclo di vita, soprattutto dopo che il DM è già entrato nel mercato e fa parte della pratica clinica routinaria.
Il sistema di vigilanza e la rete nazionale di vigilanza
Il Sistema di Vigilanza sui dispositivi medici persegue l’obiettivo di garantire un elevato livello di protezione e tutela della salute e sicurezza dei pazienti, degli operatori, degli utilizzatori, e tutti i soggetti che a vario titolo interagiscono con dispositivi, riducendo la possibilità che lo stesso tipo di incidente si ripeta.
Tale sistema, attraverso un costate monitoraggio, deve essere in grado di identificare rapidamente ogni nuovo problema/criticità legato a un dispositivo medico e di individuare quindi l'azione correttiva più idonea per eliminare o ridurre tale problematica.
Gli utilizzatori (operatore sanitario o utilizzatore profano) contribuiscono in maniera importante al raggiungimento degli obiettivi del Sistema di Vigilanza, rivestendo un ruolo chiave nella segnalazione degli incidenti rilevati durante l’ordinario utilizzo di un dispositivo.
Per favorire lo scambio tempestivo e capillare delle informazioni riguardanti gli incidenti e le azioni di sicurezza che coinvolgono i dispositivi medici, il Ministero della Salute ha istituito, con proprio decreto del 31 marzo 2022, una rete nazionale per la dispositivo-vigilanza. La rete è costituita dallo stesso Ministero (in qualità di autorità competente), dai Responsabili Regionali della vigilanza (RRV), dai Responsabili Locali della vigilanza (RLV) e dagli operatori sanitari.
Le indicazioni operative fornite con la circolare ministeriale del 29 novembre 2022 supportano l’operatore sanitario, il paziente e l’utilizzatore profano nella gestione dei modi e delle tempistiche delle segnalazioni relative agli incidenti gravi, gli incidenti non gravi e i reclami.
La rete regionale di vigilanza
La Regione Friuli Venezia Giulia ha disciplinato sul proprio territorio la rete della dispositivo-vigilanza, in coerenza con quanto disposto dal decreto ministeriale 31 marzo 2022, istituendo una rete regionale che è stata formalizzata con la delibera della giunta regionale n. 2040/2022, successivamente aggiornata con la delibera giuntale n. 2056/2023.
Obiettivo della rete regionale è di contribuire, attraverso un sistema omogeneo e organizzato di vigilanza, a migliorare la qualità delle prestazioni di un dispositivo, la sicurezza del paziente, dell’operatore sanitario e del caregiver e complessivamente garantire un’assistenza più efficiente.
La rete regionale di dispositivo vigilanza è strutturata in 2 livelli organizzativi che lavorano
in maniera integrata, assicurando un'efficace sinergia tra farmacisti, ingegneri clinici e altre
figure professionali coinvolte nel processo, coordinandosi con il servizio per la gestione del
rischio clinico:
• livello regionale, nell’ambito della Direzione Centrale Salute (DCS) in cui operano i
Responsabili regionali della vigilanza (RRV);
• livello locale, nell’ambito delle Aziende Sanitarie e degli IRCCS del SSR, in cui operano i
Responsali locali della vigilanza (RLV), e di ARCS.
I responsabili della vigilanza sui DM
Responsabili regionali della vigilanza (RRV) presso la Direzione centrale Salute, politiche sociali e disabilità:
dott.ssa Aba Pettinelli
ing. Sara Zanchiello
vigilanza.dm@regione.fvg.it
Responsabili locali della vigilanza (RLV) presso gli Enti del Servizio Sanitario
Regionale:
AZIENDA SANITARIA UNIVERSITARIA GIULIANO ISONTINA (ASUGI)
dott. Marco Cristiani -
marco.cristiani@asugi.sanita.fvg.it - 0481 585203
sostituto: dott.ssa Erika Blanco -
dispositivo.vigilanza@asugi.sanita.fvg.it - 0481 592880
Per gli IVD dott.ssa Francesca Sirianni -
francesca.sirianni@asugi.sanita.fvg.it - 040 3992431 - 339 8705278
sostituto: dott. Francesco Fontana -
francesco.fontana@asugi.sanita.fvg.it - 0481 487640
AZIENDA SANITARIA UNIVERSITARIA FRIULI CENTRALE (ASUFC)
dott.ssa Roberta Mozzon -
roberta.mozzon@asufc.sanita.fvg.it - 0432 559299
sostituto: dott.ssa Barbara Solazzo -
barbara.solazzo@asufc.sanita.fvg.it - 0432 921419
per gli IVD dott. Daniele Nigris -
daniele.nigris@asufc.sanita.fvg.it - 0432 552323
sostituto: dott.ssa Cristina Dreossi -
cristina.dreossi@asufc.sanita.fvg.it - 0432 554003
AZIENDA SANITARIA FRIULI OCCIDENTALE (ASFO)
dott.ssa Laura Cadelli -
laura.cadelli@asfo.sanita.fvg.it - 0434 399853
sostituti: dott.ssa. Chiara Biasinutto -
chiara.biasinutto@asfo.sanita.fvg.it
dott.ssa Claudia Sommaro -
claudia.sommaro@asfo.sanita.fvg.it - 0434 399467
per gli IVD dott.ssa Margherita Morandini -
margherita.morandini@asfo.sanita.fvg.it - 3387313269
IRCCS "BURLO GAROFOLO" DI TRIESTE
dott.ssa Marta Paulina Trojniak -
marta.trojniak@burlo.trieste.it - 040 3785415
sostituto: dott.ssa Anna Arbo -
anna.arbo@burlo.trieste.it
IRCCS "CENTRO DI RIFERIMENTO ONCOLOGICO" DI AVIANO
dott.ssa Antonella Bertola -
antonella.bertola@cro.it - 0434
659129
sostituto: dott.ssa Sara Cecco -
scecco@cro.it - 0434 659837
Cosa, come e quando segnalare
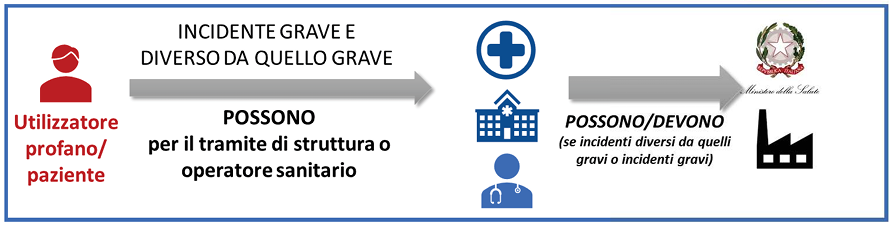
I Regolamenti Europei distinguono gli incidenti in due fattispecie: incidente grave e incidente diverso dall’incidente grave.
L’incidente grave è qualsiasi incidente che, direttamente o indirettamente, ha causato, può aver causato o può causare una delle seguenti conseguenze:
a) il decesso di un paziente, di un utilizzatore o di un'altra persona;
b) il grave deterioramento, temporaneo o permanente, delle condizioni di salute del paziente,
dell'utilizzatore o di un'altra persona;
c) una grave minaccia per la salute pubblica.
Per grave minaccia per la salute pubblica si intende un evento che potrebbe comportare un imminente rischio di decesso, un grave deterioramento delle condizioni di salute di una persona o una malattia grave che possa richiedere una tempestiva azione correttiva e che possa provocare un tasso significativo di morbilità o di mortalità umane o che è inusuale o inaspettata per quel dato luogo e momento.
L’incidente diverso dall’incidente grave è definito come qualsiasi malfunzionamento o alterazione delle caratteristiche o delle prestazioni di un dispositivo messo a disposizione sul mercato, compreso l'errore d'uso determinato dalle caratteristiche ergonomiche, come pure qualsiasi inadeguatezza nelle informazioni fornite dal fabbricante e qualsiasi effetto collaterale indesiderato; nonché, qualora trattasi di un IVD, qualsiasi danno derivante dalla decisione medica, azione od omissione basata sulle informazioni o sui risultati forniti dal dispositivo.
Gli
operatori sanitari pubblici e privati che nell’esercizio della loro attività
rilevino un incidente grave, anche solo sospetto, che veda coinvolto un
dispositivo medico, un dispositivo diagnostico in vitro (IVD), un dispositivo su misura o un
dispositivo ricompreso nell'allegato XVI del regolamento (UE) 2017/745,
sono tenuti a darne comunicazione con la massima urgenza e comunque non oltre 10
giorni dalla data in cui si è verificato l’evento:
• al Ministero della Salute compilando il modulo on line
https://nsis-ids.sanita.it/nidp/loginspid_cittadino.jsp?target=https://sisn.salute.gov.it/app/dincfe;
• al fabbricante o al suo mandatario, anche per il tramite del fornitore del dispositivo
medico, anche trasmettendo il pdf del file generato al termine della compilazione on line.

Gli
operatori sanitari pubblici e privati che rilevino
un incidente diverso da quello grave durante l’utilizzo di un dispositivo:
• devono darne comunicazione al fabbricante o al suo mandatario anche per il tramite del
distributore del DM/IVD, preferibilmente entro 30 giorni;
• possono effettuare la segnalazione d’incidente al Ministero della Salute compilando il
modulo on line,
https://nsis-ids.sanita.it/nidp/loginspid_cittadino.jsp?target=https://sisn.salute.gov.it/app/dincfe.
I Responsabili Locali della Vigilanza possono utilmente supportare l’operatore anche in questa fase.

Inoltre, l'operatore sanitario, qualora rilevi carenze correlate all'identità, qualità,
durabilità, affidabilità, usabilità, sicurezza o prestazioni del DM/IVD o relative ad un servizio
che influiscono sulle prestazioni di tali DM, è tenuto a fare segnalazione di
reclamo al Ministero della salute, tramite la compilazione del
modulo online
disponibile al link:
https://nsis-ids.sanita.it/nidp/loginspid_cittadino.jsp?target=https://sisn.salute.gov.it/app/drecfe.ù
Il reclamo deve essere inviato contestualmente anche al fabbricante, anche per il tramite del
distributore.
Il Ministero della Salute ha elaborato una linea d'indirizzo per la segnalazione dei reclami sui DM e IVD per fornire chiarimenti e indicazioni agli operatori sanitari, agli utilizzatori profani e ai pazienti.
Gli operatori sanitari pubblici o privati, nonché le strutture sanitarie, le farmacie, i medici di medicina generale o i pediatri di libera scelta che ricevono le segnalazioni del reclamo da parte degli utilizzatori profani e dei pazienti, trasmettono, entro trenta giorni, tali segnalazioni al Ministero della salute.
Qualunque altra persona, sia un paziente che un utilizzatore profano, qualora rilevi un incidente grave, un incidente non grave durante l’utilizzo di un dispositivo, può svolgere un ruolo importante nella Rete di Vigilanza sui dispositivi medici comunicandolo al Ministero della Salute, anche per il tramite dei RLV della struttura sanitaria di riferimento (si veda l’elenco responsabili aziendali della vigilanza sui DM).
Il paziente e l’utilizzatore profano (intesi come un soggetto che non possiede qualifiche formali in un ambito pertinente dell'assistenza sanitaria o in una disciplina medica) possono inoltre segnalare eventuali reclami al fabbricante anche per il tramite dei relativi operatori economici.
Le immagini riportate in questa sezione sono tratte da: Minella D, Campanale A, Ventimiglia M, Serino L, Mattei D, Goffredo R, Romano S, Rossetti A, Caddeo A, Rivitti AG, Scagliola I, Colliardo A, Croce D, Ippoliti G, Mennini F.S. Iachino A.- Rapporto di Vigilanza sui dispositivi medici - anno 2023, Ministero della salute, 2023
Protocollo regionale per lo svolgimento delle attività di vigilanza sui dispositivi medici e i dispositivi medici in vitro
La rete regionale di vigilanza ha redatto un Protocollo regionale per la gestione omogena delle
attività di vigilanza sul territorio, tenuto conto delle disposizioni normative e delle circolari
ministeriali in materia. Sono così definite le modalità organizzative nell’ambito del Servizio
sanitario regionale relativamente a:
• segnalazione degli incidenti gravi e degli incidenti diversi da quelli gravi e la gestione
dei dispositivi coinvolti;
• segnalazione dei reclami;
• diffusione degli avvisi di sicurezza;
• gestione delle azioni correttive di sicurezza.
Il Protocollo è stato adottato con atto del Direttore centrale competente in
materia.